非洲猪瘟病毒(ASFV)致死率近100%,其结构蛋白p22功能未知,阻碍疫苗研发;病毒通过降解宿主I型干扰素受体(IFNAR1)逃逸天然免疫。
2025年7月16日,河南农大动医学院与哈兽研国家非洲猪瘟专业实验室研究团队在PLOS PATHOGENS上发表题为“The African swine fever virus p22 inhibits the JAK-STAT signaling pathway by promoting the TAX1BP1-mediated degradation of the type I interferon receptor”的最新研究论文。研究阐明了p22在ASFV复制中的生物学功能,并揭示了IFNAR 1的一种新的自噬降解机制。
| IF:5.5 中科院1区 | JCR/Q1 微生物学参考译文: 非洲猪瘟病毒p22通过促进TAX 1BP 1介导的I型干扰素受体降解抑制JAK-STAT信号通路第一作者:Haojie Ren, Yanjin Wang, Lian-Feng Li通讯作者: Hua-Ji Qiu, Gai-Ping Zhang, Su Li, Wen-Rui He |
| I 主要研究结果 |
1.KP177R基因对ASFV在猪肺泡巨噬细胞(PAM)中的复制不是必需的
作者构建ASFV-ΔKP177R缺失株,在猪肺泡巨噬细胞(PAMs)中连续传代并纯化。测序与Western blot证实KP177R被完全删除且p22蛋白不表达。透射电镜显示ΔKP177R与野生型病毒形态一致;血吸附试验及生长曲线表明,缺失株的吸附能力与复制动力学与野生型无显著差异。因此,KP177R基因对ASFV在PAMs中的形态发生和复制并非必需。
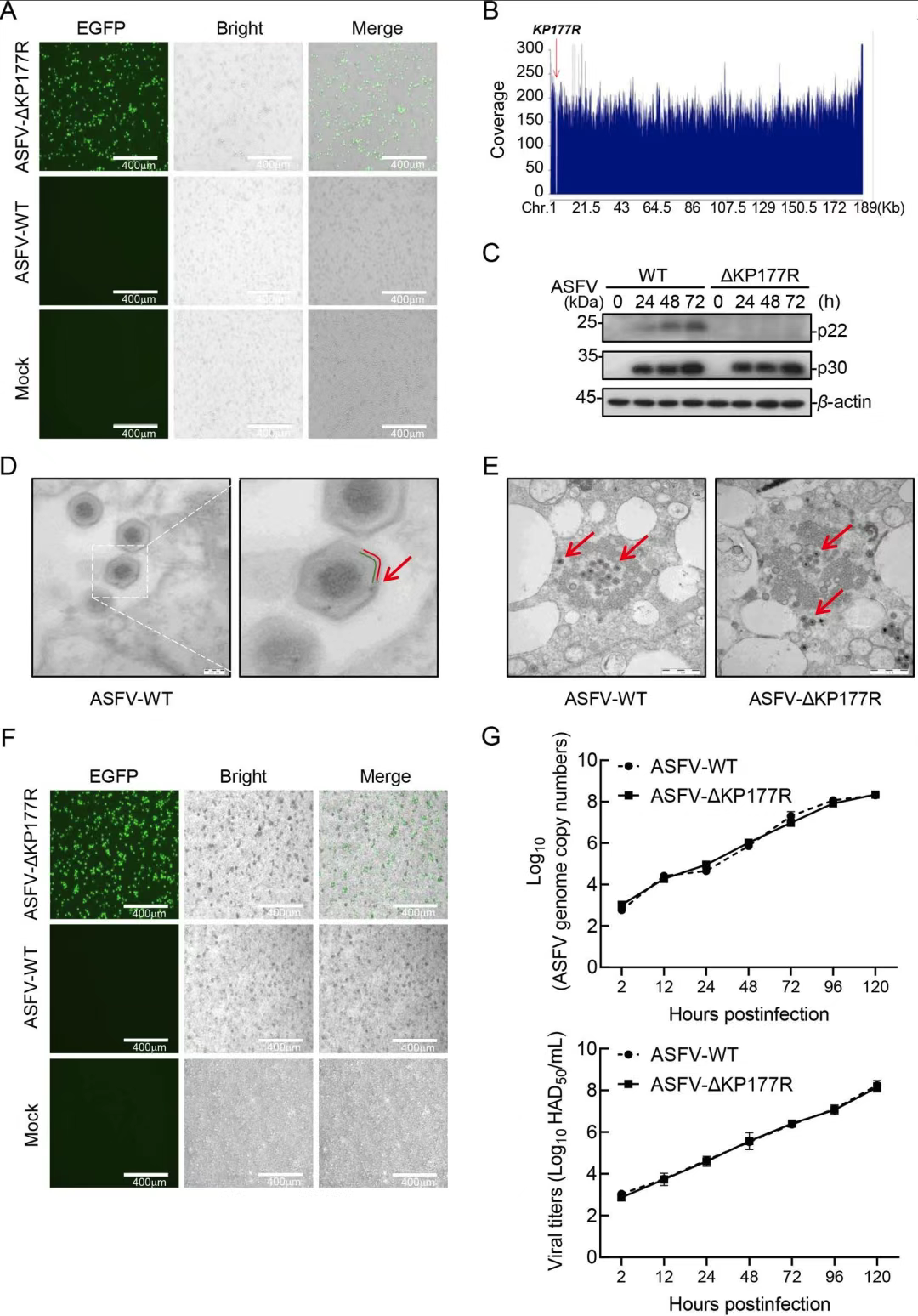
图1.KP177R基因对于ASFV在猪肺泡巨噬细胞(PAM)中的复制不是必需的
2.KP 177 R缺失的ASFV突变体显著促进PAM中JAK-STAT信号通路的激活
研究发现,与野生型ASFV(ASFV-WT)相比,缺失KP177R基因(编码p22蛋白)的突变株(ASFV-ΔKP177R)显著增强了猪肺泡巨噬细胞(PAMs)中JAK-STAT通路的激活。RNA-seq显示,ASFV-ΔKP177R感染后,JAK-STAT相关基因及干扰素刺激基因(ISGs)表达显著上调;qPCR和Western blot证实,STAT1、STAT2磷酸化水平及ISG15、IFIT1、RSAD2等转录水平均显著升高,表明p22通过抑制JAK-STAT通路发挥免疫逃逸作用。

图2.KP 177 R缺失的ASFV突变体显着促进PAMS中JAK-STAT信号通路的激活
3.ASFV p22抑制IFN-β触发的JAK-STAT信号通路的激活
本部分研究通过外源表达p22发现,ASFV p22能以剂量依赖方式显著抑制IFN-β诱导的STAT1启动子活性、STAT1/2磷酸化及下游ISG(STAT1、IFIT1、RSAD2)转录,而对TNF-α触发的NF-κB通路无影响,证实p22特异性阻断IFN-β介导的JAK-STAT信号激活。

图3.ASFV p22抑制IFN-β触发的JAK-STAT信号通路的激活
4.ASFV p22靶向IFNAR 1并促进其降解
研究显示,ASFV p22蛋白特异性结合I型干扰素受体1(IFNAR1),并显著降低其蛋白水平,但不影响IFNAR2;p22过表达或ASFV-WT感染均可诱导内源IFNAR1降解,而缺失p22的ASFV-ΔKP177R无此效应,证实p22靶向并促进IFNAR1降解,削弱宿主干扰素信号。

图4.ASFV p22靶向IFNAR 1并促进其降解
5.ASFV p22通过自噬降解IFNAR1
研究发现p22显著降低IFNAR1蛋白水平,但不影响其mRNA表达;自噬抑制剂(CQ、3-MA、SBI)可阻断该降解过程,并恢复IFNAR1水平。机制上,p22促进LC3-II上调和p62降解,诱导自噬;ATG5/ATG7缺失亦抑制IFNAR1降解,证实p22通过经典自噬途径靶向降解IFNAR1。
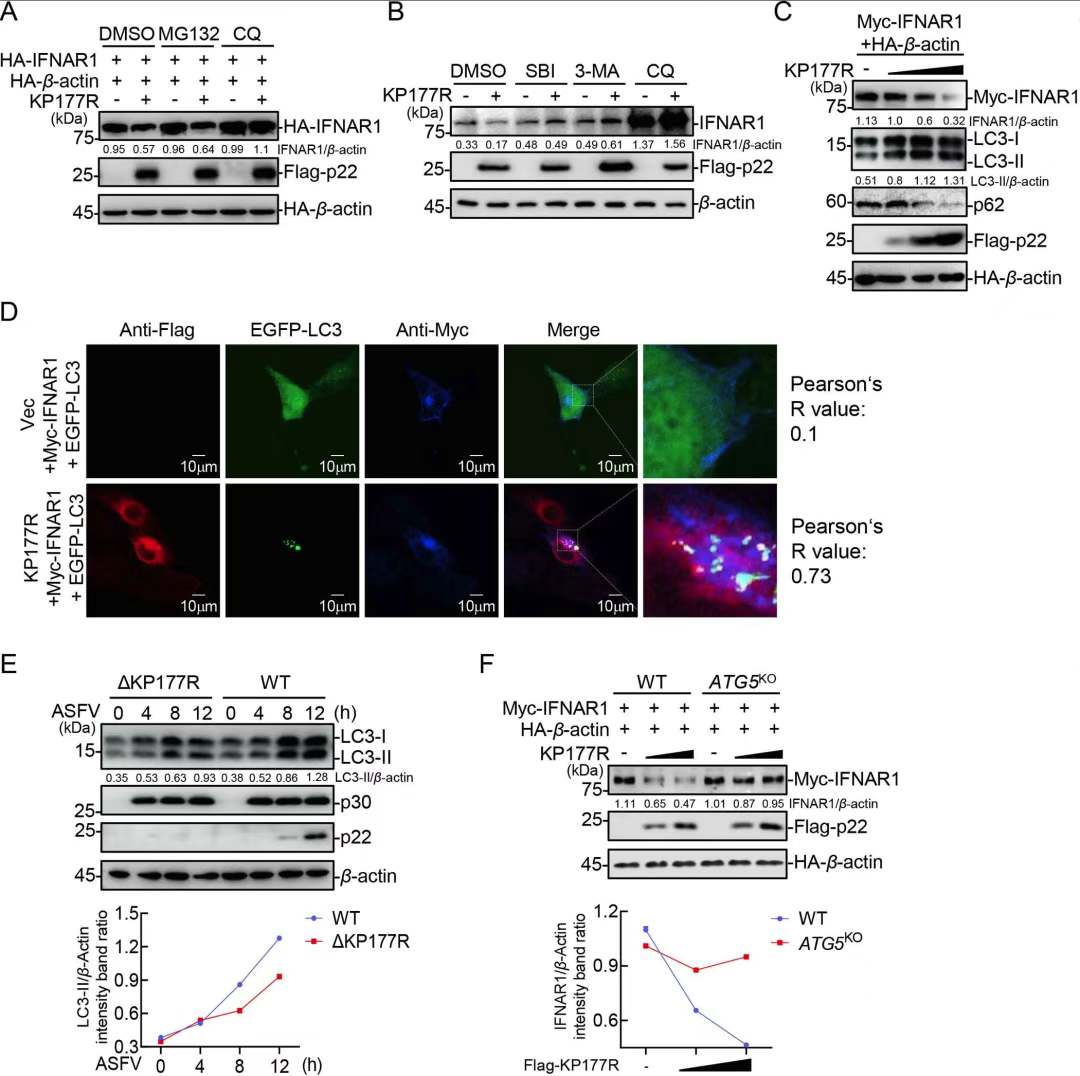
图5.ASFV p22通过自噬降解IFNAR 1
6.TAX1BP1参与p22诱导的IFNAR1降解
研究发现p22特异性结合自噬受体TAX1BP1,TAX1BP1敲除完全阻断p22介导的IFNAR1降解,并导致IFNAR1蛋白水平显著升高。机制上,TAX1BP1直接结合IFNAR1,p22通过其跨膜域增强TAX1BP1-IFNAR1相互作用,促进IFNAR1经自噬途径降解,且该过程不依赖TAX1BP1的泛素结合域。

图6.TAX1BP1参与p22诱导的IFNAR1降解
7.ASFV p22增强TAX1BP1介导的IFNAR1降解
ASFV p22通过其跨膜结构域增强TAX1BP1与IFNAR1的结合,显著加速TAX1BP1介导的自噬降解,降低IFNAR1蛋白水平。该作用独立于TAX1BP1的泛素结合域,且不依赖IFNAR1泛素化,最终抑制JAK-STAT通路。

图7.p22增强TAX1BP1介导的IFNAR1降解
8.ASFV p22的跨膜结构域对于抑制JAK-STAT信号通路的激活是必需的
研究发现,仅保留跨膜结构域(TMD)的p22突变体即可有效结合IFNAR1和TAX1BP1,促进IFNAR1自噬降解,显著抑制IFN-β诱导的STAT1/2磷酸化及下游ISGs表达;缺失TMD则完全丧失此功能,证实TMD是p22阻断JAK-STAT通路的关键区域。

图8.ASFV p22的跨膜结构域对于抑制JAK-STAT信号通路的激活是必需的
| I 研究总结 |
研究首次阐明ASFV p22通过其跨膜结构域(TMD)同时结合宿主I型干扰素受体IFNAR1与选择性自噬受体TAX1BP1,增强二者相互作用并驱动IFNAR1经自噬途径降解,从而在病毒感染早期阻断JAK-STAT信号通路的激活,抑制抗病毒基因表达。
机制上,这一过程独立于IFNAR1泛素化及TAX1BP1的泛素结合域,依赖经典自噬核心组分ATG5/ATG7。KP177R缺失病毒(ASFV-Δp22)感染猪肺泡巨噬细胞后,IFNAR1水平升高,STAT1/2磷酸化及ISGs转录显著增强,证实p22在免疫逃逸中的关键作用。
综上,p22-TAX1BP1-IFNAR1轴揭示了ASFV利用宿主自噬系统削弱天然免疫的新策略,为解析ASFV致病机制和设计靶向IFN通路的新型疫苗与抗病毒药物提供了理论依据。
源论文链接: https://doi.org/10.1371/journal.ppat.1013319




发表评论 取消回复